Our physics-driven AI delivers precise, high-accuracy results—speeding up drug development with greater efficiency.
Thank you for your interest in TQchem—your partner in accelerating drug discovery through computational chemistry.
TQchem is our proprietary computational framework, offering a unique set of modelling capabilities for chemistry and computer-aided drug design, like conformer search, molecular, and protein-protein docking. It harnesses Terra Quantum's physics-driven tensor train optimization methods and GPU-accelerated simulation techniques for exceptional performance.
Depending on the project needs, TQchem offers a variety of molecular description methods with different cost-to-accuracy ratios, including force-fields, semiempirical and ab initio methods. For large-scale calculations with high accuracy, we provide GPU-accelerated DFT code with hybrid functionals.

Our physics-driven AI delivers precise, high-accuracy results—speeding up drug development with greater efficiency.
Thank you for your interest in TQchem—your partner in accelerating drug discovery through computational chemistry.
Accelerated Conformer Search
Small Molecule Docking to Proteins
Protein-Protein Docking
GPU-Accelerated Electronic Structure
Explore a modular suite of high-performance quantum chemistry methods designed for accuracy, flexibility, and speed—tailored to meet the unique demands of your molecular modeling workflows.
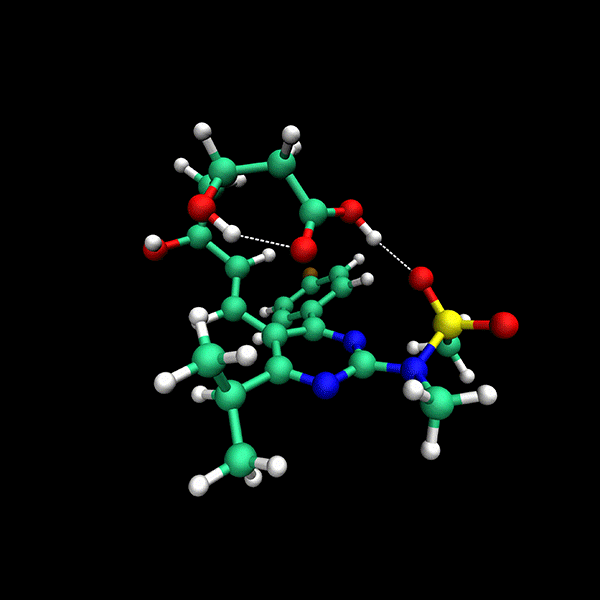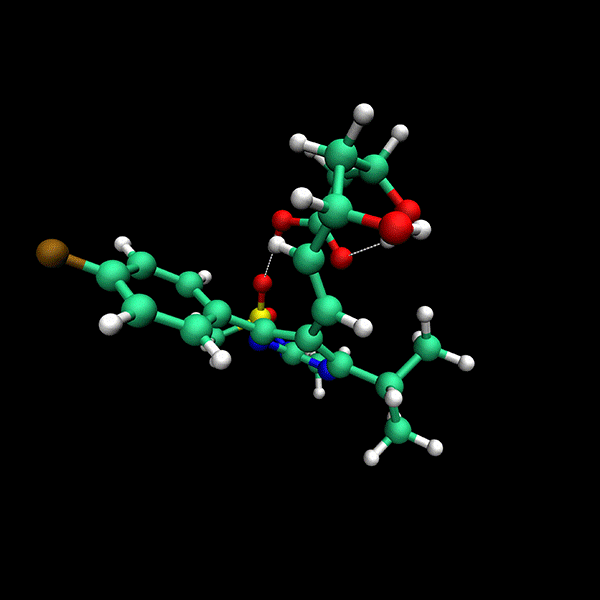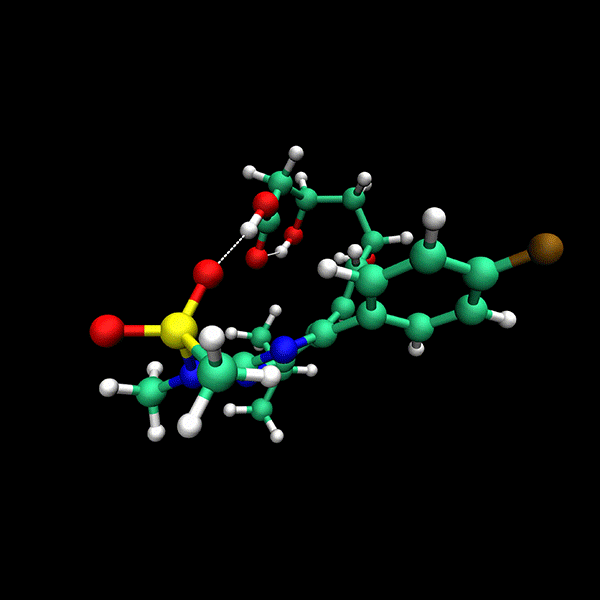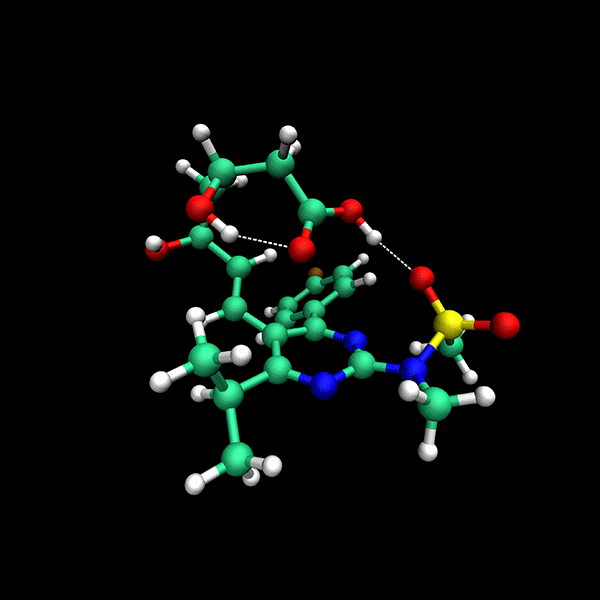
Significant Speed-Up: Provides up to 24x faster results compared to state-of-the-art methods on datasets like CD25, BACE and Astex.
High Accuracy: Delivers precise conformers with fewer function evaluations.
Supports multiple backends for energy evaluation: Force Fields, Semiempirical Electronic Structure Methods, Ab Initio Electronic Structure Methods.
Benefits:
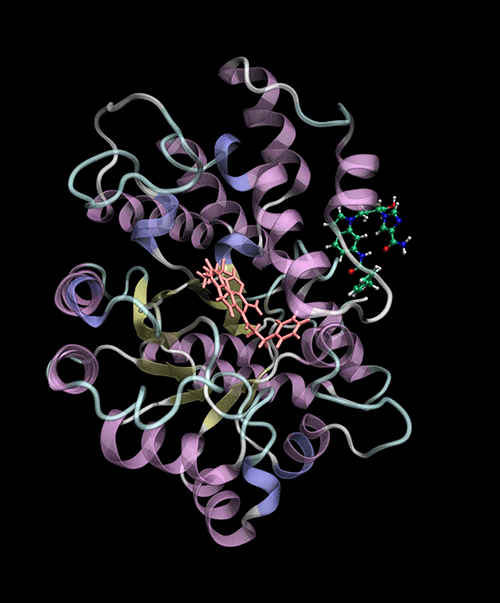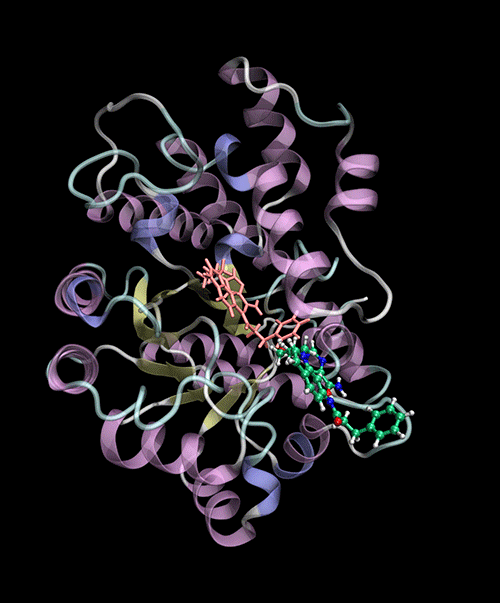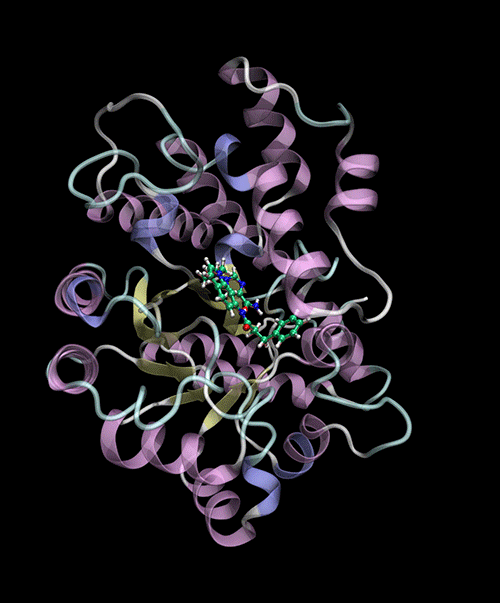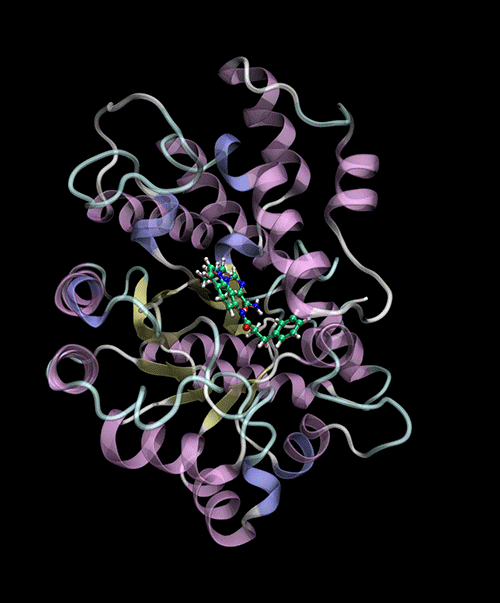
Optimized Binding Positions: Utilizes tensor train optimization to find optimal ligand binding positions.
Automatic Binding Site Detection: Identifies binding sites without prior knowledge.
Supports multiple backends for energy evaluation: Force Fields, Semiempirical Electronic Structure Methods.
Smart Optimization: Features intelligent internal coordinates to enhance optimization efficiency.
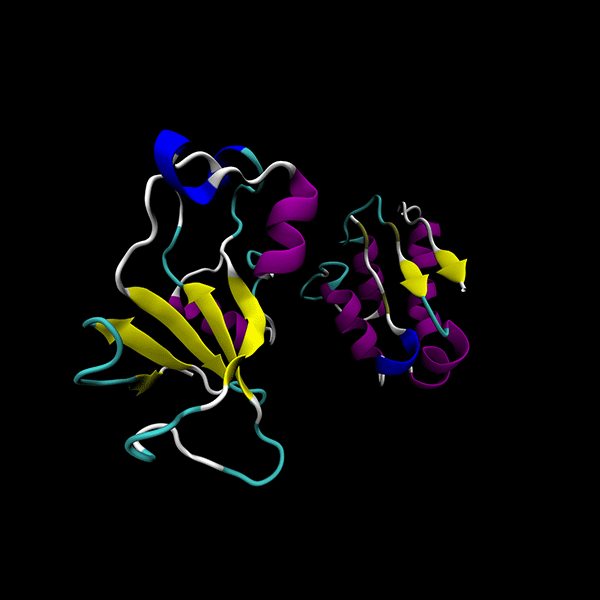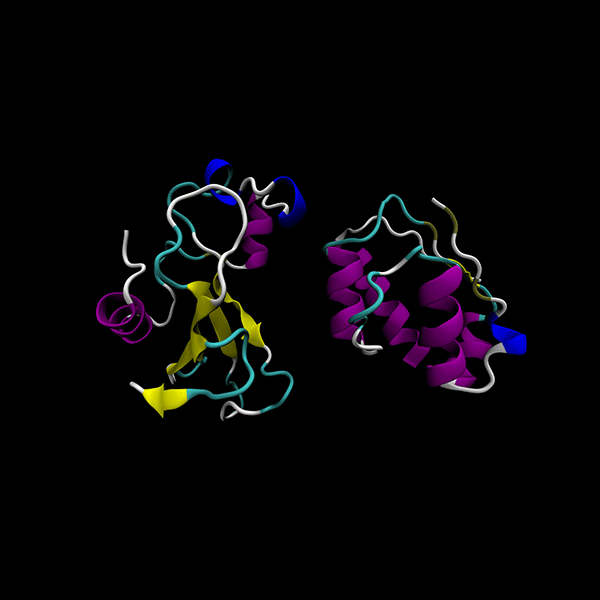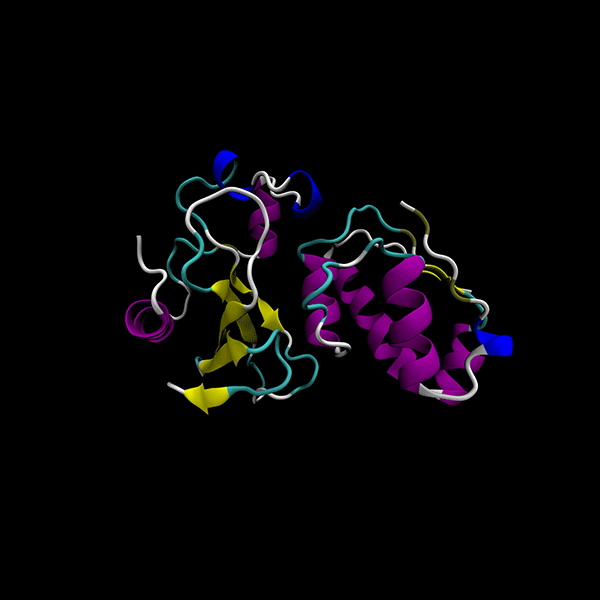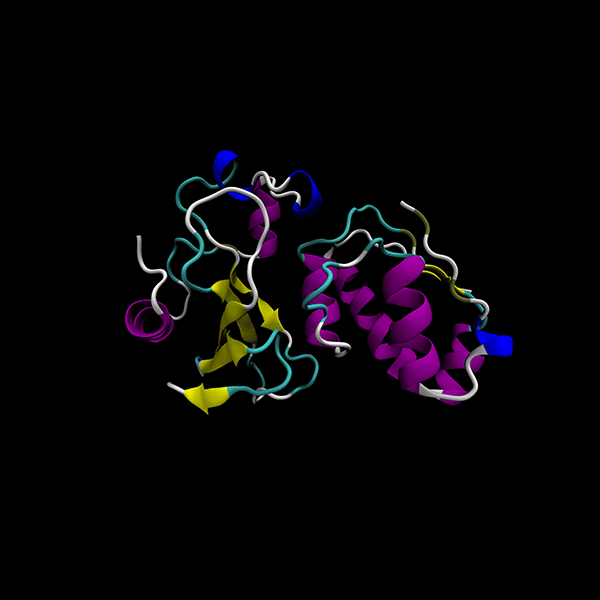
Enhanced Precision: Achieves 40% better minima than the GSO algorithm in LightDock, with comparable computational cost.
Focused on rigid-body protein-protein docking for improved performance.
Utilizes data-agnostic tensor train optimization
Supports multiple backends for energy evaluation: Force fields, Semiempirical electronic structure methods
Incredible Speed: Offers up to 400× speed-up compared to state-of-the-art CPU methods.
High Accuracy: Delivers precise energy calculations using ab initio methods.
Key Features
Advanced quantum tools for simulating and optimizing molecular structures.
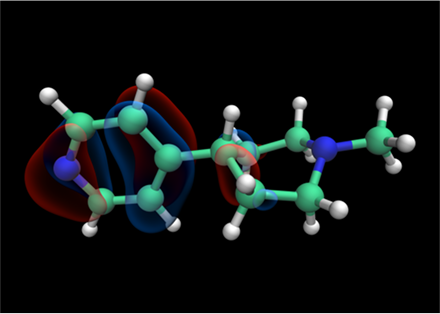
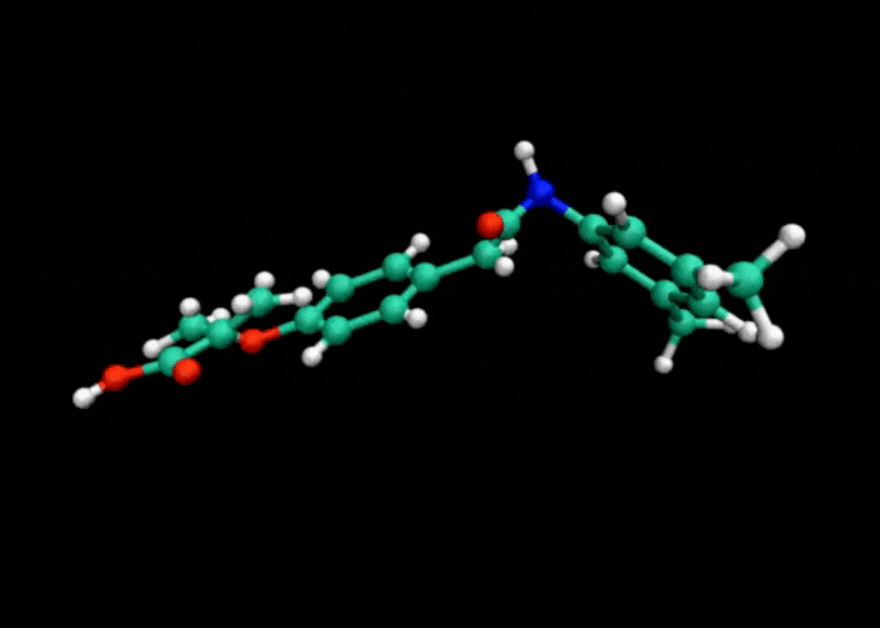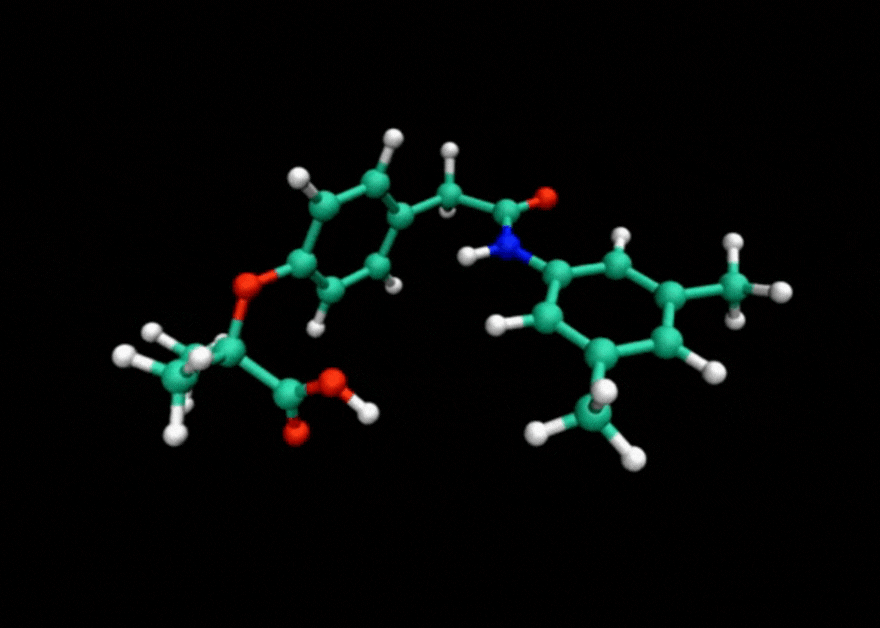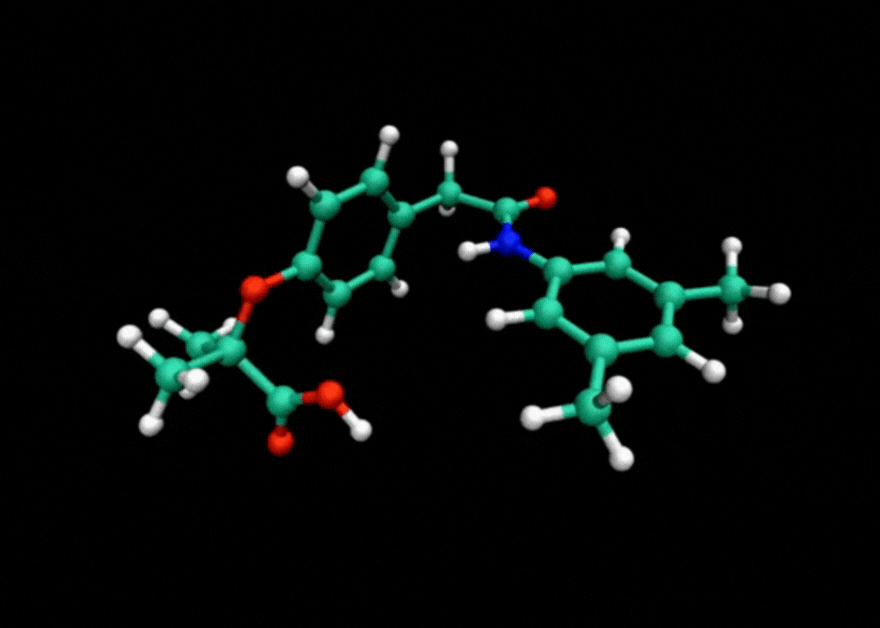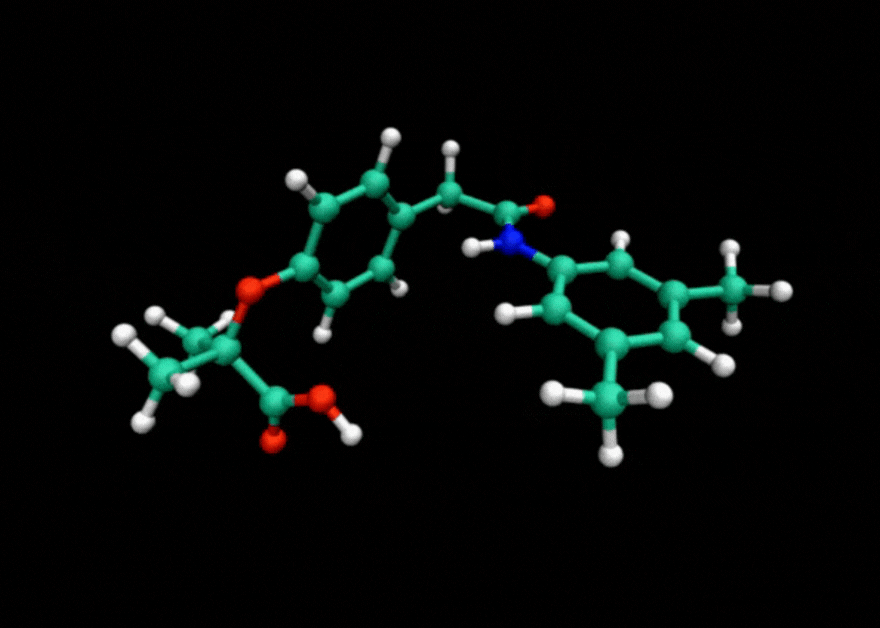
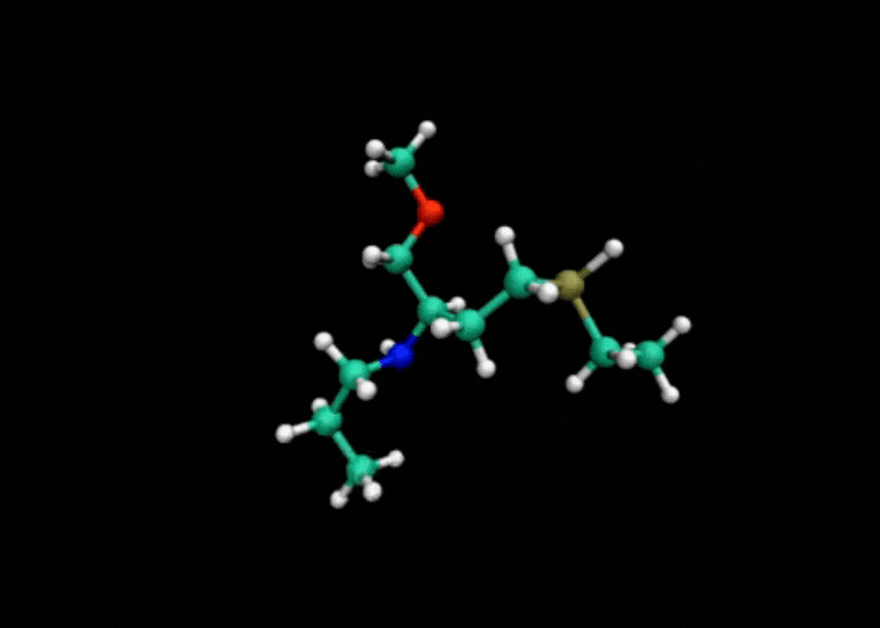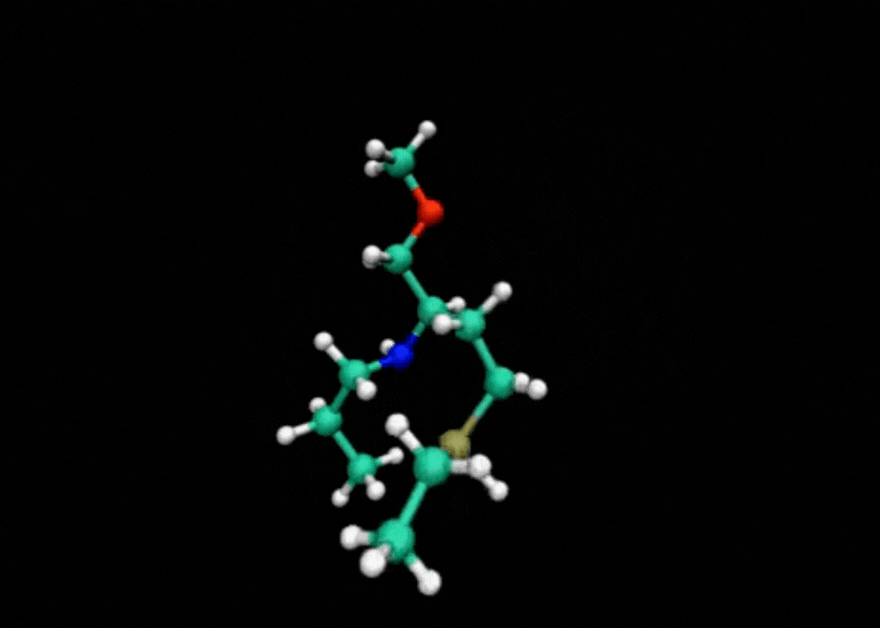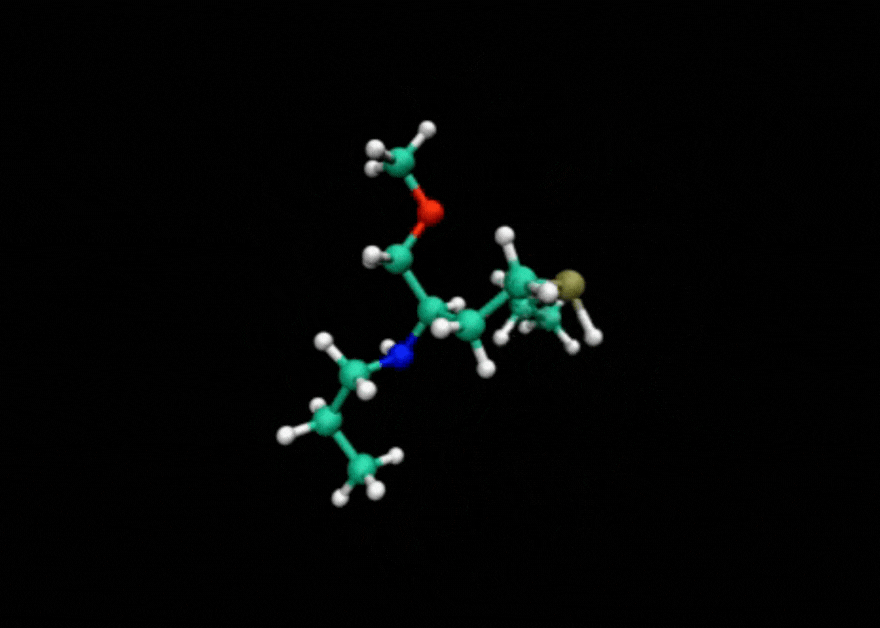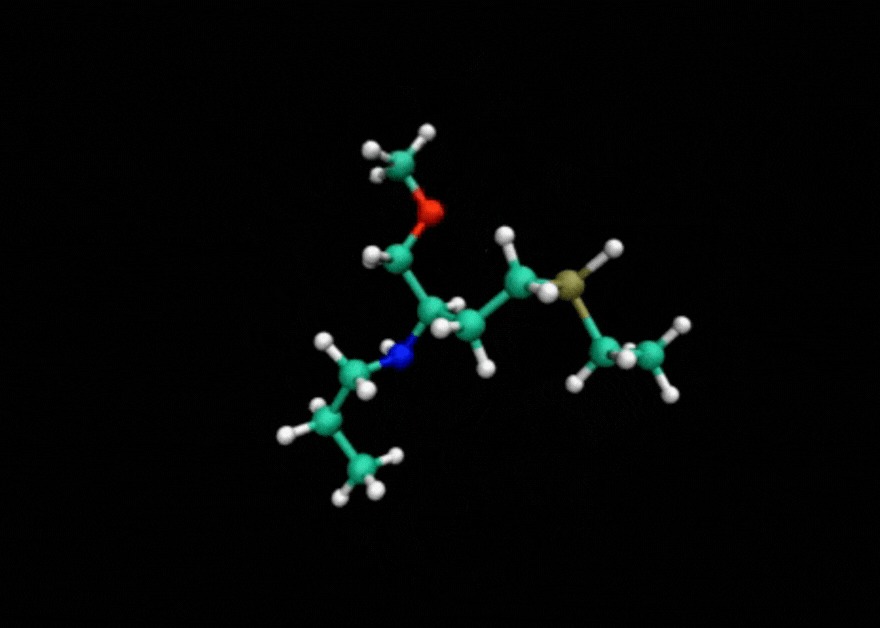

Tensor train techniques for conformer search offer a physics-driven approach, unlike many machine learning-based methods that rely on large datasets. Our method finds highly accurate conformers with significantly fewer function evaluations than competing optimization techniques, delivering substantial speed-ups and a data-independent solution.

Terra Quantum supports academic and non-commercial research by providing free access to selected components. If you are an academic researcher or involved in non-commercial work, we invite you to apply for a license key to explore our advanced computational chemistry tools. Please reach out to our team to learn more and obtain your license.
Terra Quantum is committed to supporting research progress, offering free access to selected features of TQChem. If you'd like to experiment with TQchem for non-commercial projects, we invite you to apply for a non-commercial license key to explore TQchem’s advanced computational chemistry tools.
Developed in close collaboration with the academic community
Features an API to allow easy integration to the existing pipelines
Features separate Python API and web interfaces to run experiments
Explore groundbreaking findings and real-world quantum applications.





















Terra Quantum is committed to supporting research progress, offering free access to selected features of TQChem. If you'd like to experiment with TQchem for non-commercial projects, we invite you to apply for a non-commercial license key to explore TQchem’s advanced computational chemistry tools.
Our physics-driven AI delivers precise, high-accuracy results—speeding up drug development with greater efficiency.
Thank you for your interest in TQchem—your partner in accelerating drug discovery through computational chemistry.